Conformational change coupled to ligand binding
A ten-state kinetic model showing that the balance between conformational selection and induced fit is set by ligand concentration — not fixed by the protein.
THE CHALLENGE
Conformational selection, or induced fit?
Molecular recognition carries a long-running debate. Does a ligand bind a high-affinity shape the protein already samples on its own — conformational selection, binding after folding — or does it bind first and pull the protein into shape — induced fit, binding before folding? The two are usually argued as mutually exclusive.
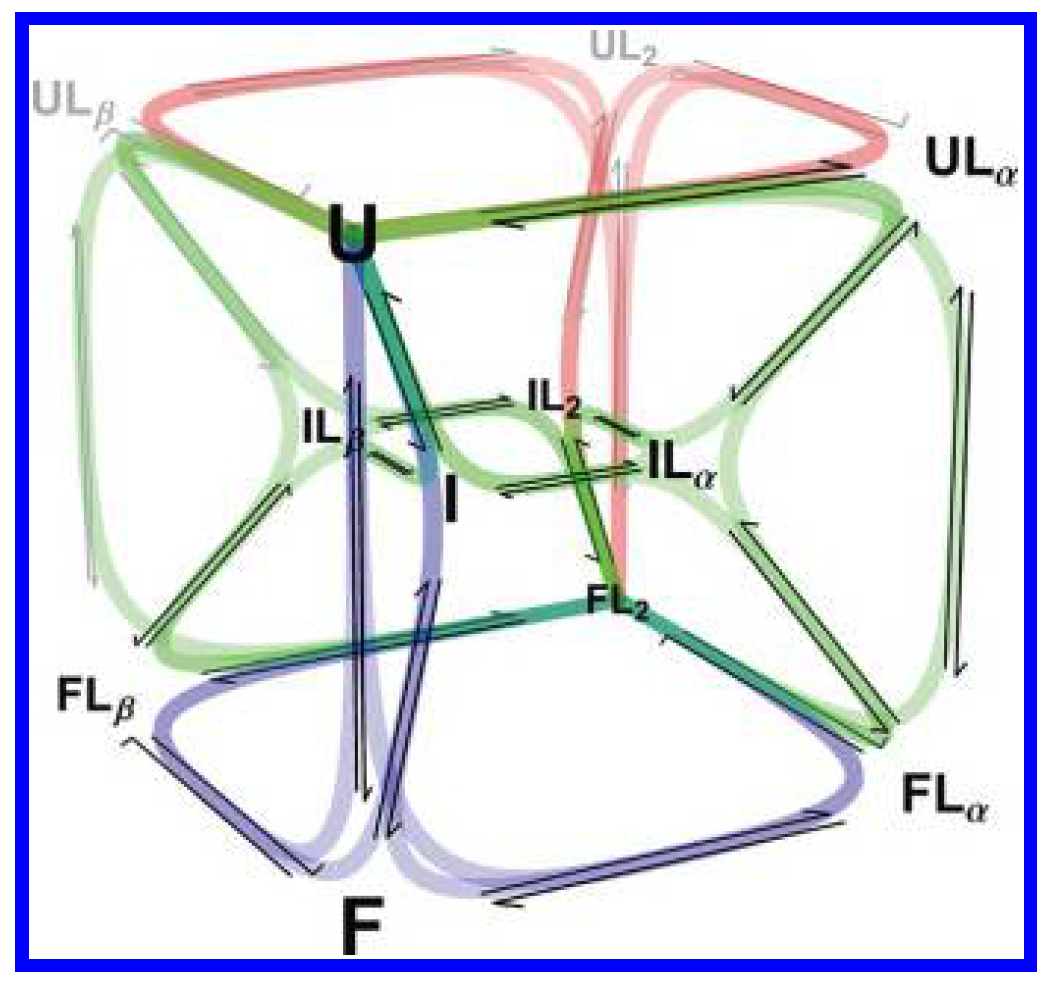
RNase P's P protein is an ideal place to settle it: folding and pyrophosphate (PPi) binding are tightly coupled, with ligand able to bind at multiple sites across multiple folding states.
WHAT WE DID
One model, fit to everything at once
We built a ten-state kinetic network — every combination of folding state and ligand-bound state — and fit it globally across stopped-flow experiments spanning a range of ligand concentrations. Every rate constant was constrained to be consistent across all conditions simultaneously, so the mechanism had to explain the full dataset, not one curve at a time.
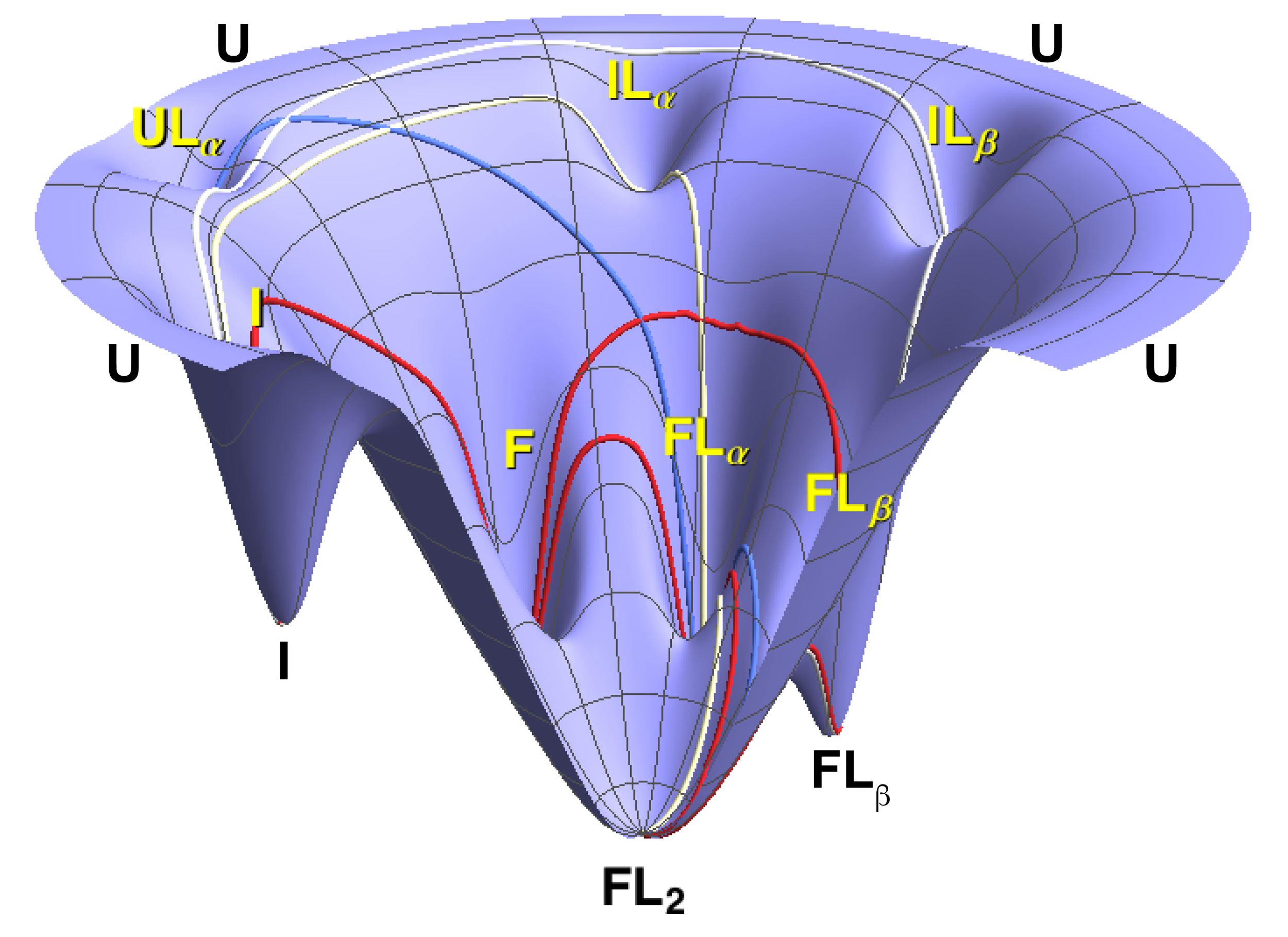
Because the fit returns every microscopic rate constant, we could compute the fractional flux through all 18 folding-and-binding pathways as a function of [PPi] — turning “which mechanism?” into a direct, per-pathway accounting.
WHAT IT REVEALED
A mixed mechanism tuned by ligand concentration
Flux is continuously partitioned between the two mechanisms, with pyrophosphate concentration setting the balance. At low [PPi], ~90% of it runs through conformational selection — the protein folds on its own and PPi binds the already-folded state. Near the apparent Kd, the two run side by side; well above it, induced-fit and mixed routes take over. The mechanism isn't a fixed property of the protein, nor a binary choice between the two — it's a continuously shifting balance set by the conditions.
Conformational selection and induced fit aren't rival answers — they're two ends of a continuum, and ligand concentration sets where you sit on it.
Antibody-binding domains fold independently
A steep N-to-C stability gradient across five independently folding domains.
Read case study →Exposing hidden high-affinity states
A 6-state model: the highest-affinity state is present all along, hidden by its low population.
Read case study →Have a system like this?
Tell us about the protein, the data, and the question. We'll tell you whether a kinetic or thermodynamic model can answer it.
Contact Us→